
Um bei Blutgerinnungsstörungen medizinisch zu intervenieren, muss man die Funktionsweise der Gerinnung genau verstehen. Welche Faktoren die Gerinnung verursachen und welche sie hemmen können, erfahren Sie hier.
Als Hämostase bezeichnet man die Mechanismen, die schrittweise die Blutung stoppen. Der hämostatische Prozess wird in zwei verschiedene Phasen unterteilt: Blutstillung (primäre Hämostase) und Blutgerinnung (sekundäre Hämostase).
- Erhöhte Blutungsneigung und verminderte Blutgerinnung = Hämorrhagische Diathese
- Erhöhte Thromboseneigung = Thrombopathie
- Störungen der primären Hämostase
- Störungen der sekundären Hämostase = Störung der plasmatischen Gerinnung oder der Fibrinolyse
Primäre Hämostase
Darunter versteht man die vorläufige rasche Blutstillung innerhalb weniger Minuten (ca. 1-3 Minuten) durch Vasokonstriktion der glatten Muskulatur der am Verletzungsort vorhandenen Blutgefäße unter Bildung eines so genannten weißen Thrombus/Gerinnsels, der die Blutungsstelle zunächst einmal bedeckt und so abdichtet.
Zur Vasokonstriktion der Blutgefäße an der Verletzungsstelle kommt es durch ein Zusammenziehen der glatten Muskeln der Gefäße und der Aktivierung bestimmter Nerven durch die Verletzung selbst– zusätzlich werden bestimmte Mediatoren wie Serotonin, Katecholamine, Plättchenfaktoren etc. aus dem Gewebe (Endothel) und aus den Blutplättchen freigesetzt. Die Thrombozyten spielen, obgleich sie die kleinsten Zellen des Blutes sind, eine entscheidende Rolle bei der primären Hämostase. Im strömenden Blut sind sie an den Endothelwänden – also am Rand der Blutströmung – konzentriert und können so rasch bei lokalen Endothel - Verletzungen reagieren bzw. mit einbezogen werden.
Im Falle einer Verletzung kommt es zu einem Anheften der Thrombozyten an die Gefäßwand. Dies wird vermittelt durch den von-Willebrand-Faktor (vW-Faktor) – dieser bindet sich einerseits an das verletzte Endothel (Kollagen!) und andererseits an sogenannte Adhäsionsmoleküle auf den Thrombozyten (z.B. GP Ib/IX; GP Ia/IIa). Weitere Blutplättchen binden sich dann mittels GP IIb/IIIa über Fibrinogen an schon festhaftende Thrombozyten und aneinander (Aggregation). Dadurch wird im Inneren der Thrombozyten das freie Kalzium erhöht, das die Adhäsion, Aggregation und Degranulation von Mediatoren aus den Granula der Thrombozyten zusätzlich steigert.
Bei einer Verletzung der Gefäßwand wird aus Phospholipiden der Thrombozytenmembran Arachnoidonsäure abgespalten – daraus entstehen die beiden Gegenspieler:
- Thromboxan A2 → Vasokonstriktion und Aggregation der Thrombozyten
- Prostazyklin (aus Endothelzellen) → Hemmung der überschießenden Plättchenaggregation und Vasodilatation.
Therapeutisch gibt man Acetylsalicylsäure (ASS), um die Bildung von Thromboxan A2, also die Vasokonstriktion, zu hemmen. Thromboxan A2 wird z.B. verstärkt gebildet beim Rauchen plus Kontrazeption mit der "Pille". Das Risiko für Thrombosen steigt!
Größere zusammenhängende Blutplättchenaggregate entstehen hauptsächlich über die zunächst reversible Bindung des GP-Rezeptors IIb/IIIa – über Fibrinogen zu anderen Thrombozyten und deren Rezeptor.
Sekundäre Hämostase
Zu ihr kommt es während der nächsten 6-10 Minuten nach Beginn einer Blutung mit der Aktivierung von Pro-Faktoren der Blutgerinnung, die im Blutplasma in inaktiver Form vorliegen. Durch deren Aktivierung kommt es so schließlich zur Ausbildung eines endgültigen, stabilen Verschlusses der Wunde – durch einen sogenannten "gemischten Thrombus". Er besteht aus einem Fibringerüst und den darin eingeschlossenen Blutzellen (Thrombozyten, Erythrozyten und Granulozyten). Danach wird der Defekt schließlich wieder "fest" verschlossen (Epithel, Bindegewebe etc.) und der Thrombus schließlich wieder aufgelöst (=Fibrinolyse). Nachdem durch die primäre Hämostase nur ein vorläufiger Verschluss einer Wunde (innen oder von außen) durch einen Thrombozyten-Pfropf erreicht wurde, muss diese schließlich dauerhaft verschlossen werden. Geschähe dies nicht, wären Nachblutungen bzw. erneute Blutungen die Folge. Dies nennt man sekundäre Hämostase bzw. "endgültige Blutstillung".
Zahlreiche Gerinnungsfaktoren, die im Plasma als inaktive Vorstufen vorliegen, aktivieren sich gegenseitig durch enzymatische Spaltung oder sind Ko-Faktoren bei diesen Reaktionen. "Aktiv" werden sie durch Abspaltung von Proteinen (=Peptiden) in Anwesenheit von Ca-Ionen. Die Komplexe, die sich dabei bilden, lagern sich an verletzten und damit offen zugänglichen Endothelanteilen (z.B. Kollagen) an, auch an interstitiellen Geweben, an Zellmembranen von Thrombozyten und verschiedener Gewebe.
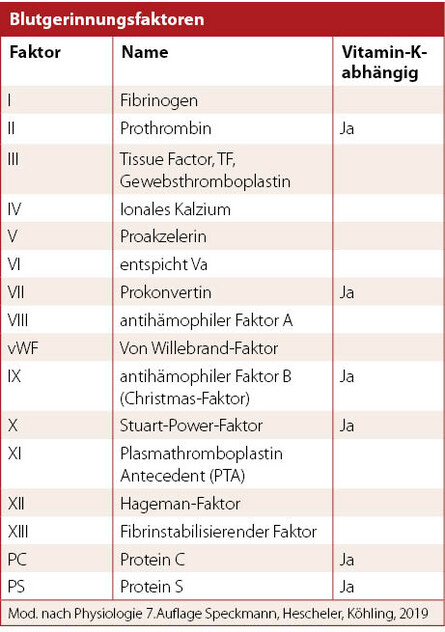
Merke: Das Endprodukt der Blutgerinnung ist ein Netzwerk aus Fibrin, das dem gebildeten Thrombus seine Festigkeit verleiht. Die einzelnen Gerinnungsfaktoren werden mit römischen Ziffern bezeichnet (I – XIII), wobei ein nachgestelltes kleines "a" für den aktivierten Gerinnungsfaktor (z.B. ii a) steht.
Start und Verlauf
Ein entscheidender Faktor ist der Tissue Faktor (TF =Gewebefaktor), als ein Eiweiß normaler Bestandteil von Zellmembranen, die üblicherweise keinen Kontakt mit dem strömenden Blut haben (z.B. in der Adventitia der Blutgefäße). Nach seiner Aktivierung kann dieser auch von Endothelzellen, Monozyten und Thrombozyten abgegeben werden. Über die Aktivierung von Faktor VII in Anwesenheit von Kalzium startet jetzt die Gerinnungskaskade (=Exogenes/Extrinsic-System). Über den aktivierten Faktor X (=Xa) kommt es schließlich zur Bildung von Thrombin. Durch einen positiven Rückkopplungsmechanismus mit anderen Gerinnungsfaktoren (VIII, V, XI und IX) kommt es zur weiteren und anhaltenden Thrombinbildung, was schließlich zu einem stabilen Gerinnsel führt. (siehe Abbildung).
Ende durch abschließende Bildung eines "roten Thrombus"
Am Ende des extrinsic und auch des intrinsic Weges steht das Thrombin – dieses Thrombin spaltet schließlich das langkettige Fibrinogen in die Peptide A und B. Diese löslichen Fibrin-Monomere polymerisieren schließlich zu Fibrin und bilden so faserige Strukturen, die das Plasma quasi "gelieren" lassen, unter Einverleibung von Gewebeanteilen in der unmittelbaren Umgebung. Durch den Faktor XIII werden diese Strukturen schließlich verfestigt und stabilisiert (= Fibrinstabilisierender Faktor). Thrombasthenin aus den Blutplättchen selbst bewirkt schließlich ein Zusammenziehen der Wundränder – die Wunde ist fest verschlossen!
Merke: Ein weißer Thrombus entsteht bei einer sehr langsamen Gerinnung, wenn die roten Blutzellen (Erythrozyten) quasi vorher "absacken".
Die Auflösung von Blutgerinnseln, die Thrombolyse
In dem Moment in dem ein Thrombus entsteht, startet auch schon gleichzeitig ein System, das eine überschüssige Reaktion verhindern soll. Aktivatoren und Proaktivatoren aus den Endothelzellen – der wichtigste ist das tPA (=tissue Plasminogen-Aktivator) und die Pro-Urokinase werden freigesetzt.
Pro-Urokinase wird durch Kontaktaktivierung bei der Gerinnung zu aktiver Urokinase umgewandelt. Diese fördern die Umwandlung von inaktivem Plasminogen in Plasmin, welches vom Fibrinogen Bruchstücke abspaltet, so genannte "Fibrinspalt-Produkte" welche laborchemisch im Rahmen der Lyse (auch bei der Therapie!) nachgewiesen werden können. Damit auch die Fibrinolyse nicht überschießend reagiert, wird diese gleichzeitig auch wieder gehemmt (z.B. durch PAI 1 (=Plasminogen activator inhibitor), C1 Inhibitor).
Im Körper vorhandene "natürliche" Hemmer der Blutgerinnung
- Antithrombin III (=AT III) ist der wichtigste Gerinnungshemmer. Er bindet mit Thrombin und Faktor Xa sowie weiteren Gerinnungsfaktoren und bildet so einen Thrombin-Antithrombin-Komplex und verhindert damit eine überschießende Thrombinbildung.
- Protein S und C sind weitere wichtige Vitamin-K-abhängige Hemmer der Blutgerinnung. Ein Mangel führt zu einem erhöhten Thromboserisiko.
- Der Tissue factor pathwayinhibitor ist ein "physiologischer" Hemmer von Faktor Xa.
Wo werden die Gerinnungsfaktoren gebildet?
Die Gerinnungsfaktoren II, VII, IX und X, aber auch Protein C und S werden in der Leber hergestellt und sind von Vitamin K als Ko-Faktor abhängig (für die γ-Carboxylierung). Wenn diese Vitamin-K-abhängigen Faktoren nicht gebildet werden können oder in zu geringer Konzentration besteht eine erhöhte Blutungsneigung. Z.B. bei:
- Schweren Lebererkrankungen
- Parenteraler Ernährung - /Mangelernährung
- Einer Fettresorptionsstörung (Vit. K = fettlöslich)
- Einsatz von Vitamin-K-hemmenden Substanzen, z.B. Marcumar®, Falithrom®, Coumadin® zur Hemmung der Blutgerinnung
Wenn einzelne Gerinnungsfaktoren fehlen, kann dies ebenfalls zu einer gesteigerten Blutungsneigung führen – z.B. bei den "Blutern" – bei denen ein Mangel der Faktoren VIII (A) oder IX (B) zur Hämophilie A oder B führen. Ein Faktor XI-Mangel kann ebenfalls das Blutungsrisiko steigern.
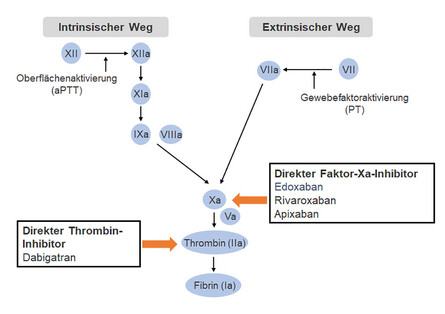
Gerinnungskaskade: Verfügbare Nicht-VKA-orale Antikoagulanzien (NOAK).
Welche Laboruntersuchungen sind wann sinnvoll?
Die verschiedenen Aktivierungsschritte der Blutgerinnung laufen im Labor bei der In-vitro-Untersuchung über zwei alternative Kaskaden ab.
Der endogene Weg (Intrinsic-System= langsam ablaufende Gerinnung)
beginnt über den Faktor XII und Kontakt mit fremden Oberflächen im Komplex (z.B. endogen an kollagenen Fasern des Endothels!) – und führt über die Faktoren XII, XI, IX und Faktor VIII schließlich zur Bildung des Faktor X, wo sich der endogene und der exogene Weg wieder treffen.
Der endogene Weg und auch die gemeinsame Endstrecke der Gerinnungskaskade können mittels der aktivierten partiellen Thrombinzeit (aPTT) im Labor überprüft werden!
Der Exogene Weg (Extrinsic-System =schnell ablaufende Gerinnung)
verläuft nur über einen Aktivierungsschritt: Gewebsthromboplastin (TF=Tissue-Faktor, Faktor III) bindet bei Gewebeverletzungen den aktivierten Faktor VIIa und aktiviert so schließlich den Faktor X, wo sich endogener und exogener Weg auch wieder treffen. Der Tissue-Faktor ist der einzige transmembranöse Gerinnungsfaktor, der nur in geringen Mengen im Blut zirkuliert. Die Überprüfung des exogenen Weges und der gemeinsamen Endstrecke erfolgt über den Quick-Test (=Prothrombinzeit, Thromboplastinzeit) – sie sind gleichwertige Tests.
Über die Aktivierung von Faktor X münden beide Wege in eine gemeinsame Endstrecke der Blutgerinnung.
Das leisten Gerinnungstests
Die Gerinnung kann mit speziellen Tests überprüft werden:
- 1. INR (=international normalized ratio). Diese liegt normalerweise bei 0,5-1,15. Der Vergleich erfolgt mit einem Normalplasma (international sensitivity index) [INR = Thromboplastinzeit des Patienten geteilt durch Thromboplastinzeit einer Kontrolle]. Der Quicktest zur Beurteilung der Effektivität bei einer Behandlung mit Cumarinen zur Antikoagulation ist stets von der eingesetzten Thromboplastincharge (Labor) abhängig – deshalb gibt man die Thromboplastinzeit besser als INR an.
- 2. Quick-Test (=Thromboplastinzeit/Prothrombinzeit): Der exogene (Aktivierung von Faktor III durch Tissue-faktor) und der gemeinsame Weg der Blutgerinnung können so getestet werden. Der Normwert liegt bei 11-15 sec, das entspricht einem Quickwert von 70-125%.
- 3. aPPT (= aktivierte partielle Thromboplastinzeit). Sie liegt normalerweise bei 25-38 sec. und dient der Überprüfung des endogenen Weges (z.B. auch bei Hämophilie A und B, Faktor XII, XI, IX und VIII). Sie reagiert besonders empfindlich auf eine Heparin-Therapie.
- 4. Thrombinzeit: Sie liegt bei 16-20 sec. und bewertet speziell die Zeit zur Bildung von Fibrin aus Fibrinogen. Sie dient auch als Suchtest bei Gerinnungsstörungen, zur Überwachung der Therapie mit unfraktionierten Heparinen und erfasst auch die Fibrinolyse (Labor und Diagnose, Thomas, 6. Aufl.)
|
|

Erschienen in: Diabetes-Forum, 2024; 36 (4) Seite 12-15